

Abstract
Background
Sickle cell disease (SCD) is a progressively debilitating genetic disease causing significant morbidity and early mortality for which a universal curative therapy is lacking. Expression of an anti-sickling β-globin via gene transfer into hematopoietic stem cells (HSCs) may reduce or eliminate SCD symptoms. LentiGlobin Drug Product (DP) contains autologous CD34+ cells transduced with the BB305 lentiviral vector (LVV) encoding β-globin with an anti-sickling substitution (T87Q). The safety and efficacy of LentiGlobin in adults with severe SCD is being evaluated in the ongoing multi-center Phase 1 study HGB-206 (NCT02140554). The first 7 patients (Group A) received DP from bone marrow harvested (BMH) HSCs and demonstrated stable but sub-optimal gene therapy-derived hemoglobin (HbAT87Q). The protocol was amended to include pre-harvest transfusions, increased target busulfan levels and a refined DP manufacturing process (Group B). The study is now enrolling patients in Group C, treated under modified protocol and including DP manufactured from plerixafor-mobilized HSCs. Data from patients in fully enrolled Groups A and B are shown here.
Methods
Patients ≥18 years old with severe SCD, as previously described, were enrolled. CD34+ cells from BMH were transduced with the BB305 LVV to produce LentiGlobin DP. Following myeloablative busulfan conditioning, patients were infused with DP. Group A patients received DP from the original manufacturing process. One Group B patient (1313) received DPs from a combination of original and refined manufacturing processes, while the other (1312) was entirely from the refined process. Adverse events (AEs), engraftment, vector copy number (VCN) in peripheral blood (PB), Hb fractions, and hemolysis markers were monitored.
Results
Nine patients (7 Group A, 2 Group B) with severe SCD (median age 26 [min - max: 18 - 42] years) were treated with LentiGlobin gene therapy. DP characteristics are shown in Table 1 and were improved in Group B patients, with higher VCNs, cell doses and % transduced cells compared to Group A. As of May 15, 2018, all Group A patients had completed ≥2 years follow-up and enrolled in a long-term follow-up study. Median follow-up was 24.2 (min - max: 22.8 - 32.9) months in Group A; 14.3 and 8.5 months for Group B patients 1313 and 1312. All patients engrafted. The toxicity profile was consistent with myeloablative conditioning. Serious AEs were reported in 8 patients; vaso-occlusive pain (n=5) was most common. No grade ≥3 DP-related AEs and importantly, no evidence of graft failure, veno-occlusive liver disease, replication competent lentivirus or clonal dominance were observed.
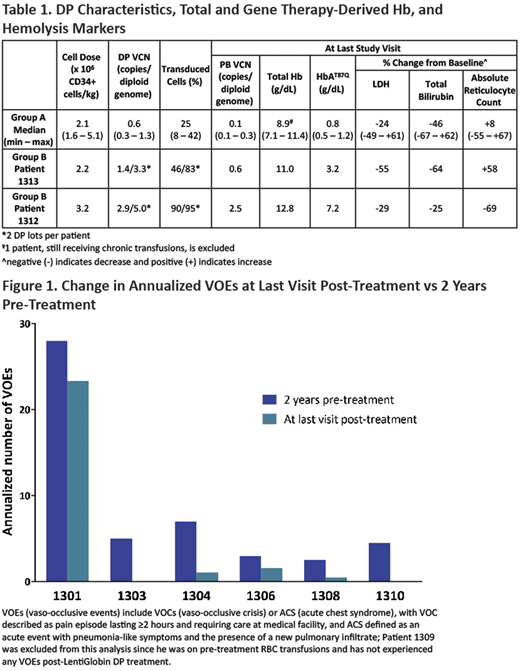
All patients demonstrate stable PB VCN and HbAT87Q levels over prolonged follow-up. In Group A, PB VCN and HbAT87Q levels were modest, with a median of 0.1 c/dg and 0.8 g/dL at last visit, respectively (Table 1). Unsupported total Hb in 6/7 patients (1 patient is on transfusions) ranged from 7.1 - 11.4 g/dL. Total bilirubin and lactate dehydrogenase (LDH) at last visit vs. baseline decreased by a median of 46% (n=7) and 24% (n=6), respectively, while reticulocyte count increased by a median of 8% (n=7). With modest HbAT87Q production the annualized vaso-occlusive events (VOEs) rate decreased 17 - 100% compared to the 2-years pre-DP infusion (n=6; Figure 1).
PB VCN and HbAT87Q in Group B patients were improved (Table 1). Total Hb at last visit was 11.0 g/dL in patient 1313 (29% HbAT87Q). In patient 1312, who received DP entirely from refined manufacturing, total Hb was 12.8 g/dL, with HbAT87Q contributing 56%. Total bilirubin and LDH normalized in both Group B patients, reticulocyte count decreased in patient 1312 (Table 1). Changes in VOE rates will be presented.
Summary
In the initial HGB-206 cohorts (Groups A and B), the safety profile of LentiGlobin gene therapy observed to date, is consistent with myeloablative busulfan conditioning. While HbAT87Q levels in Group A are sub-optimal, they are stable through ≥2 years of follow-up and most patients show a decrease in VOEs, suggesting that even modest HbAT87Q production may improve the clinical status of patients with SCD. Patients in Group B had improved DP characteristics, increased PB VCN and HbAT87Q levels with normalization of Hb in 1 patient and normalization of LDH and total bilirubin in both. Improvements in DP manufacturing correlate with increased levels of therapeutic HbAT87Q and could lead to significant clinical benefit.
Kanter:NHLBI: Membership on an entity's Board of Directors or advisory committees, Research Funding; Sancilio: Research Funding; Pfizer: Research Funding; ASH: Membership on an entity's Board of Directors or advisory committees; AstraZeneca: Membership on an entity's Board of Directors or advisory committees; Global Blood Therapeutics: Research Funding; Novartis: Membership on an entity's Board of Directors or advisory committees, Research Funding; Apopharma: Research Funding; bluebird bio: Membership on an entity's Board of Directors or advisory committees, Research Funding. Kwiatkowski:bluebird bio: Consultancy, Honoraria, Research Funding; Agios Pharmaceuticals: Consultancy, Research Funding; Apopharma: Research Funding; Terumo: Research Funding; Novartis: Research Funding. Mapara:Incyte: Consultancy. Schmidt:German Cancer Research Center: Employment; bluebird bio: Consultancy; GeneWerk GmbH: Employment. Miller:bluebird bio: Employment, Equity Ownership. Pierciey:bluebird bio: Employment, Equity Ownership. Shi:bluebird bio: Employment, Equity Ownership. Ribeil:bluebird bio: Employment, Equity Ownership. Walters:bluebird bio: Research Funding; ViaCord Processing Lab: Other: Medical Director; AllCells Inc.: Other: Medical Director; Sangamo Therapeutics: Consultancy. Thompson:La Jolla Pharmaceutical: Research Funding; Biomarin: Research Funding; Novartis: Research Funding; Celgene: Research Funding; bluebird bio: Consultancy, Research Funding; Amgen: Research Funding; Baxalta/Shire: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal